4. Generating Pickaxe Inputs
4.1. Compound Inputs
Pickaxe takes a few input files to specify compounds and rules for the expansion. One group of these files are simply compounds, some of which are required and others are option, depending on the desired functionality of a given Pickaxe run.
Required:
Compounds to react.
Optional:
Targets to filter for.
Metabolomic data to filter with (see met_data_path parameter in Built-In Filters).
4.1.1. Compound Input
Pickaxe accepts a .csv or a .tsv that consists of two columns, an id field and a structure field. The id field is used to label the final output and the structure field consists of SMILES representation of compounds.
Here is an example of a valid compound input file:
id,SMILES
glucose,C(C1C(C(C(C(O1)O)O)O)O)O
TAL,C/C1=CC(\O)=C/C(=O)O1
4.1.2. Target Input
The target compound input file takes the same form as the input compounds.:
id,SMILES
1,C=C(O)COCC(C)O
4.2. Reaction Operator Inputs
There are two files required for the application of reactions:
Reaction operators to use.
Coreactants required by the reaction operators.
Default rules are supplied with pickaxe, however custom rules can be written and used.
4.2.1. Default Rules
4.2.1.1. Overview
A set of biological reaction rules and cofactors are provided by default. These consist of approximately 70,000 MetaCyc reactions condensed into generic rules. Selecting all of these rules will result in a large expansion, but they can be trimmed down significantly while still retaining high coverage of MetaCyc reactions.
Number of Rules |
Percent Coverage of MetaCyc Reactions |
20 |
50 |
100 |
78 |
272 |
90 |
500 |
95 |
956 |
99 |
1221 |
100 |
Additionally, a set of intermediate reaction rule operators are provided as well. These operators are less generalized than the generalized ruleset and provide uniprot information for each operator.
4.2.1.2. Generating Default Rule Inputs
Default rules are imported from the rules module of minedatabase and have a few options to specify what is loaded:
Number of Rules
Fractional Coverage of MetaCyc
Anaerobic Rules only
Groups to Include
Groups to Ignore
Possible groups to ignore and include are: aromatic, aromatic_oxygen, carbonyl, nitrogen, oxygen, fluorine, phosphorus, sulfur, chlorine, bromine, iodine, halogen. Examples of Defining rules are given below.
The provided code returns the rule_list and coreactant_list that is passed to the pickaxe object.
4.2.1.3. Generalized Rules Mapping 90% Metacyc
from minedatabase.rules import metacyc_generalized
rule_list, coreactant_list, rule_name = metacyc_generalized(
fraction_coverage=0.9
)
4.2.1.4. Generalized Rules with 200 Anaerobic and Halogens
from minedatabase.rules import metacyc_generalized
rule_list, coreactant_list, rule_name = metacyc_generalized(
n_rules=200
anaerobic=True,
include_containing=["halogen"]
)
4.2.1.5. Intermediate Rules with all Halogens except Chlorine
from minedatabase.rules import metacyc_intermediate
rule_list, coreactant_list, rule_name = metacyc_intermediate(
include_containing=["halogen"],
exclude_containing=["chlorine"]
)
4.2.2. Generating Custom Rules
In the event that the default rules do not contain a reaction of interest, it is pososible to generate your own rules. Outlined below is the process to generate rules for esterification reactions, which consists of three parts
Writing the reaction SMARTS.
Writing the reaction rule.
Writing the coreactant list.
4.2.2.1. Writing Reaction SMARTS
Rules are generated using SMARTS which represent reactions in a string. Importantly, these reaction rules specify atom mapping, which keeps track of the species throughout the reaction. To higlight a simple reaction rule generation, a deesterification reaction will be used.
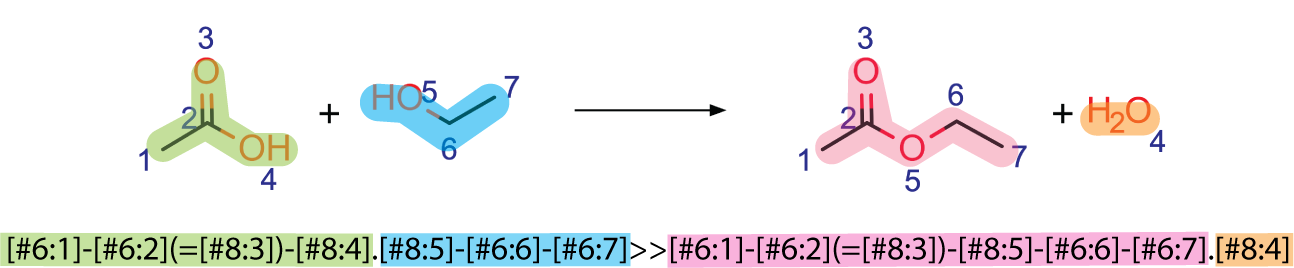
The reaction SMARTS is highighted the same color as the corresponding molecule in the reaction above. Ensuring correct atom mapping is important when writing these rules. This is an exact reaction rule and it matches the exact pattern of the reaction, which is not useful as it will not match many molecules.
Instead of using an exact reaction, a generic reaction rule can be used to match more molecules. In this case, the radius of the atom away from the reactive site is decreased.
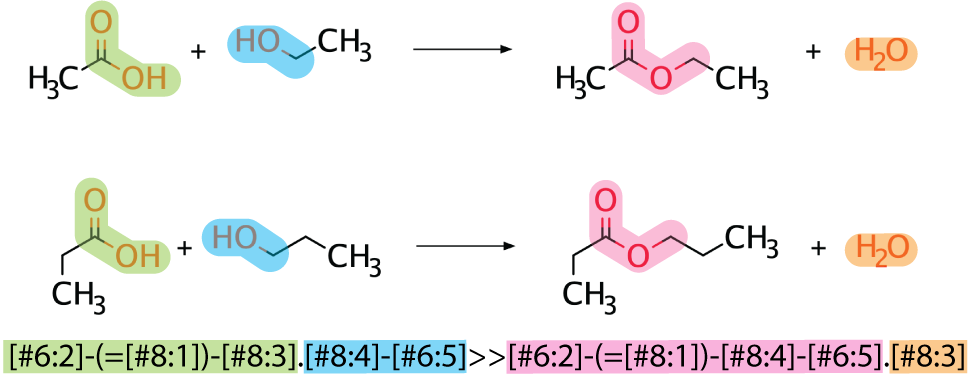
4.2.2.2. Writing Reaction Rules
With the reaction SMARTS written, now the whole rule for Pickaxe must be written. The rules are written as follows in a .tsv:
RULE_ID REACTANTS RULE PROODUCTS NOTES
The rule_id is an arbitrary, unique value, the reactants and products specify how many compounds a rule should be expecting, and the rule is the reaction SMARTS. Notes can be provided, but have no effect on the running of Pickaxe. The reactants and products are specified as a generic compound, “Any”, or as a predefined coreactant.
Below is an example of a reaction rule made for a deesterification reaction.
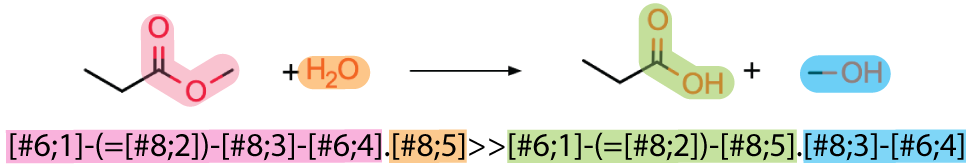
RULE_ID REACTANTS RULE PROODUCTS NOTES
rule1 Any;WATER [#6:2]-(=[#8:1])-[#8:4]-[#6:5].[#8:3]>>[#6:2]-(=[#8:1])-[#8:3].[#8:4]-[#6:5] Any;Any
Note
Currently only one “Any” is allowed as a reactant and any other reactant must be defined as a coreactant.
4.2.2.3. Defining Coreactants
Coreactants are defined in their own file that the Pickaxe object will load and use alongside the reaction rules. The coreactant file for the example deesterification reaction is:
#ID Name SMILES
WATER WATER O
4.2.2.4. Reaction Rule Example Summary
Summarized here is the input files for a deesterification reaction.
Reaction
Reaction Rule Input
RULE_ID REACTANTS RULE PROODUCTS NOTES
rule1 Any;WATER [#6:2]-(=[#8:1])-[#8:4]-[#6:5].[#8:3]>>[#6:2]-(=[#8:1])-[#8:3].[#8:4]-[#6:5] Any;Any
Coreactant Input
#ID Name SMILES
WATER WATER O